Biology is full of sequences. An organism's hereditary information is encoded as a DNA molecule, and chains of amino acids fold into proteins, the cell's major functional molecules. Comparing such sequences is at the heart of many bioinformatic analyses. For example, it enables inferring the evolutionary history of a set of sequences (e.g., genes) or linking specific mutations to phenotypes. Such analyses require sequences to be aligned, i.e., we have matched the nucleotides or amino acids sharing an evolutionary ancestor among the different input sequences.
To illustrate this, consider two seemingly different sequences ATGCTTA and TGCAATCA. We identify several shared nucleotides (|) by introducing gaps (-) and allowing mismatches (*) (Figure 1).
ATGC--TTA
||| |*|
-TGCAATCAFigure 1. Example alignment between two DNA sequences.
Experts in their domain used to painstakingly create such alignments by hand. These days, we use algorithms to compute them. Pairwise alignment algorithms compute alignments between two sequences, while multiple sequence alignment (MSA) algorithms compute alignments for a set of sequences, e.g., for a set of genes.
Partial order alignment (POA) is one type of MSA algorithm with applications in pangenome graph construction, de novo genome assembly, and many others. For example, the recently released human pangenome reference was created with an algorithm that includes POA. POA can additionally be used to error-correct reads, e.g., as a preprocessing step before de novo genome assembly.
POA represents the MSA as a directed acyclic graph (DAG) and works by iteratively computing sequence-to-graph alignments, updating the graph to include the new sequence after each alignment (Figure 2). The original algorithm introduced by Lee et al. is a dynamic programming approach, and the most recent implementation of this algorithm is called SPOA. SPOA utilizes SIMD instructions on modern CPUs, efficiently computing the dynamic programming matrix.
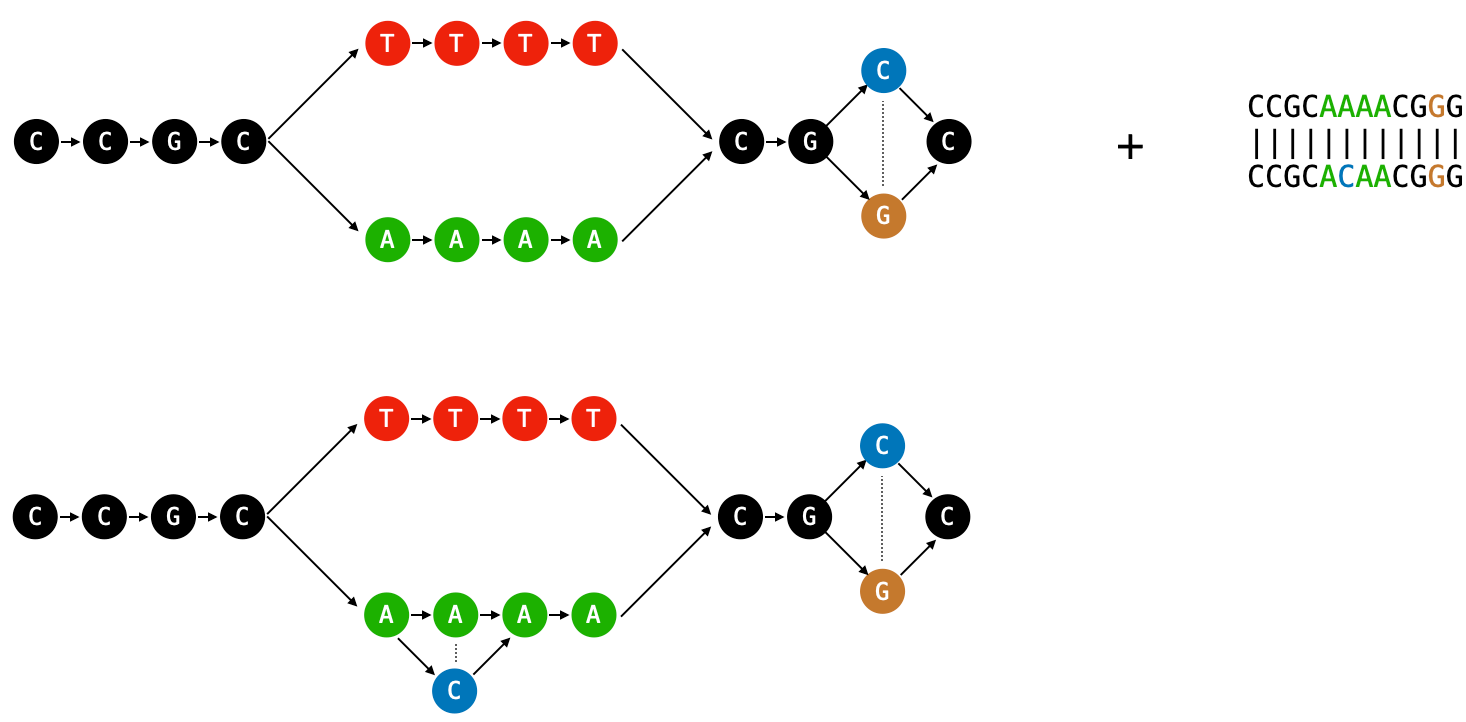
POASTA is another approach to POA. It reformulates each sequence-to-graph alignment as a mathematically equivalent "shortest path" problem. Finding shortest paths is a common computer science problem, and many existing algorithms exist. Two classic algorithms are Dijkstra's algorithm and the A* algorithm.
POASTA uses A* as its main algorithmic framework and includes three algorithmic innovations to accelerate alignment: 1) a novel A* heuristic that guides the algorithm more quickly to the target, 2) a depth-first extension component that exploits matches between the query sequence and the graph, and 3) it uses the graph topology to detect alignment states that will not be part of the optimal solution and skips those (Figure 3). These techniques substantially reduce the required computations to find the optimal alignment.
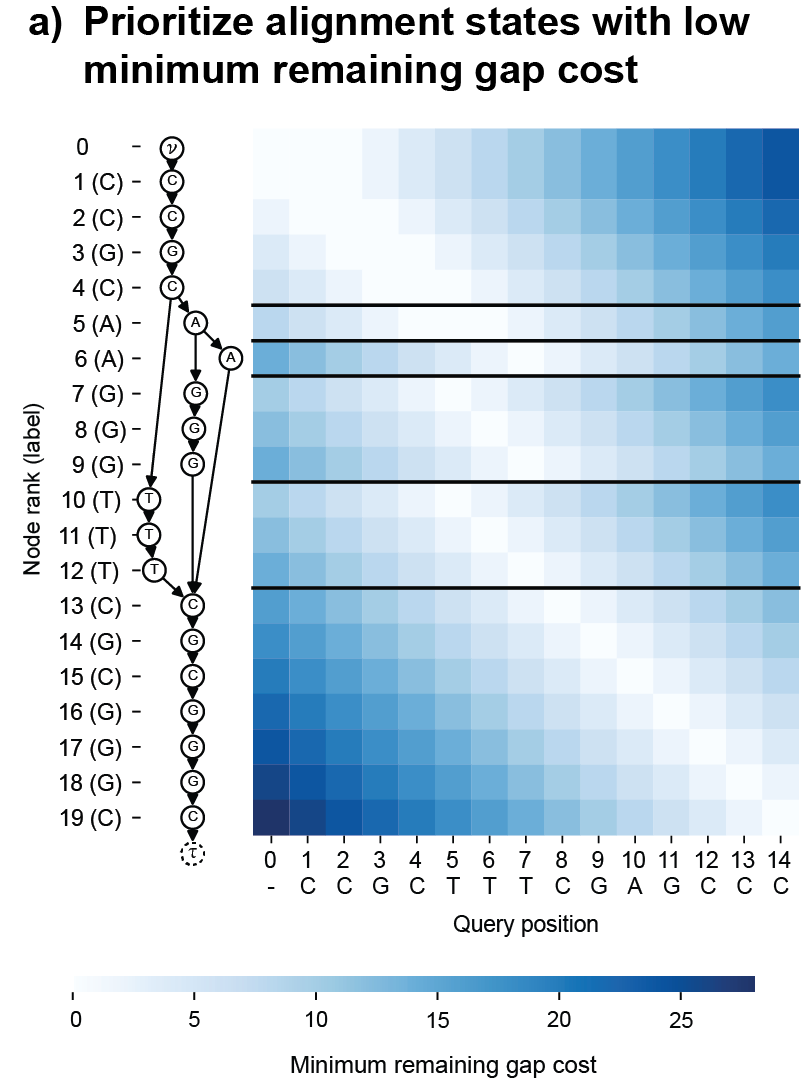
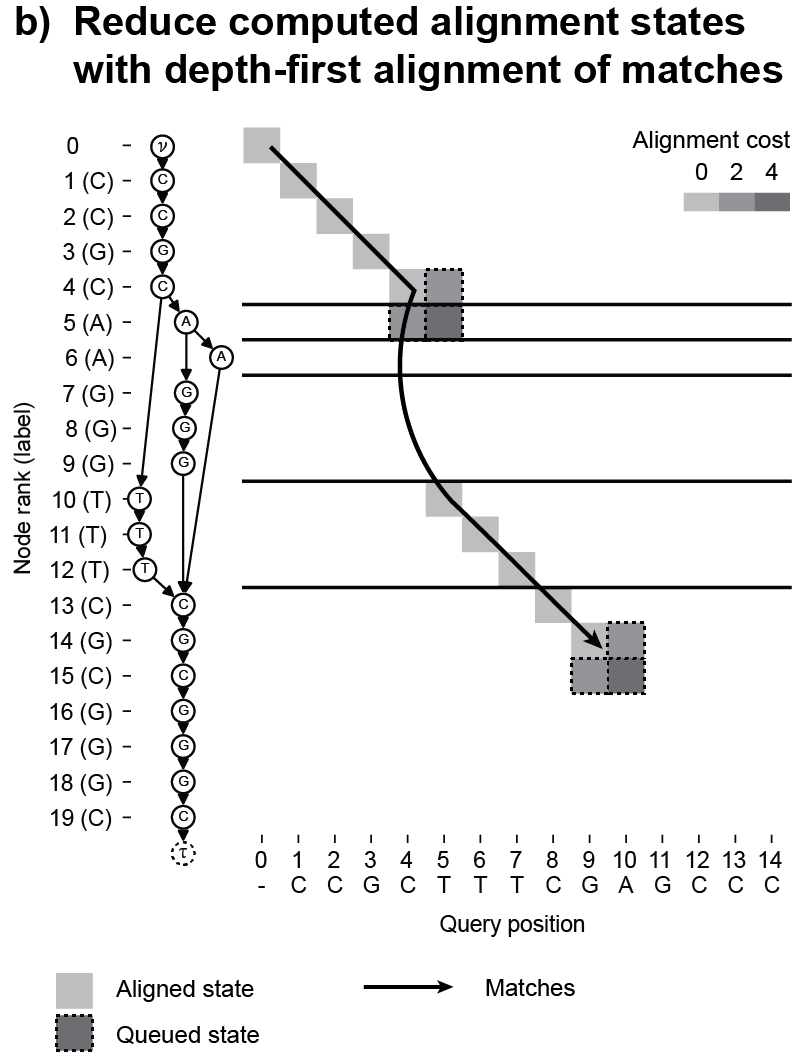
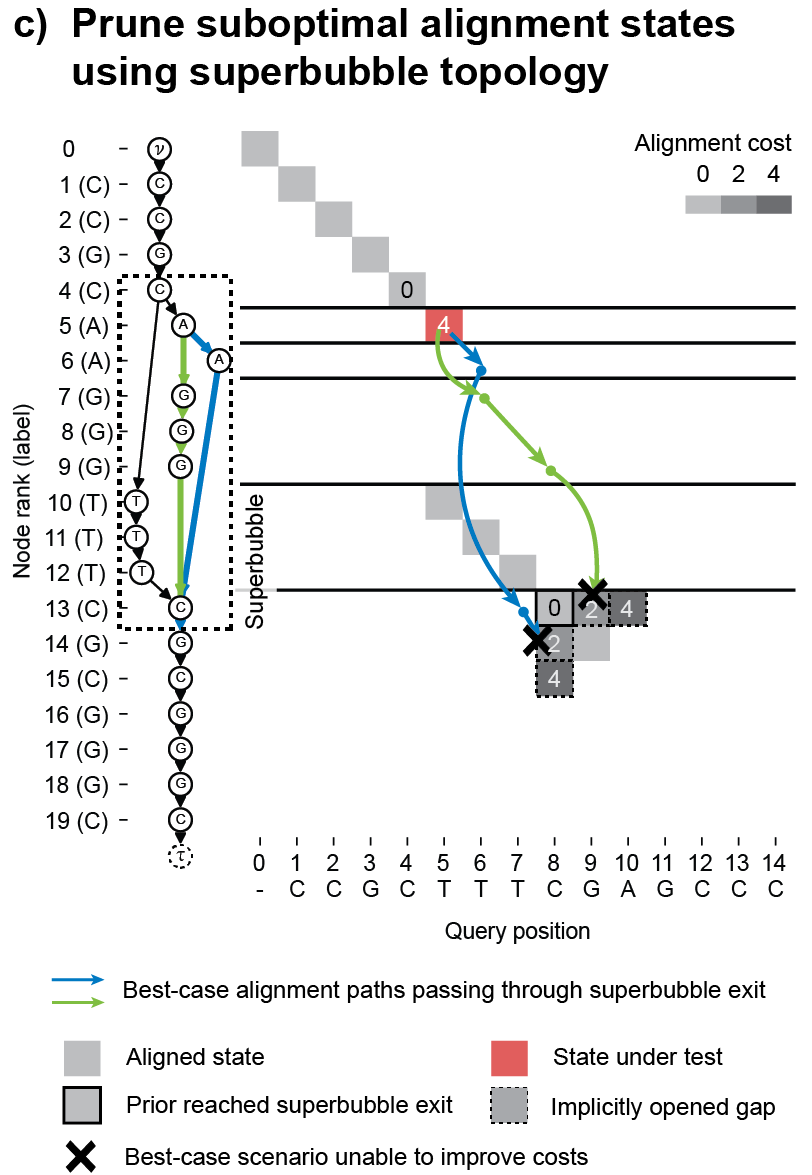
Figure 3. POASTA accelerates alignment using three algorithmic innovations. (a) POASTA introduces a "minimum gap cost" A* heuristic. (b) POASTA exploits exact matches between the query sequence and the graph in a depth-first manner. (c) POASTA uses "superbubbles" to detect and prune suboptimal alignment states.
Besides accelerating alignment, it reduces the required amount of memory, enabling alignment of longer sequences than what was possible before. In the paper, we created MSAs of 342 ~1 megabase Mycobacterium tuberculosis sequences!
POASTA is written in Rust and is available under the BSD-3 license.