We humans each carry thousands of bacterial species. They inhabit our skin, nose, gut, and several other sites. Many species protect us from pathogens, help digest food, or train our immune system. Other species, however, can cause life-threatening infections.
Even within the same bacterial species, enormous differences in pathogenicity exist. For example, almost every human harbors the bacteria Escherichia coli in their gut microbiome without any issues. Some strains, however, can cause severe diarrhea or urinary tract infections.
Because of this phenotypic diversity, it is important to know what specific strain is present when analyzing microbial communities. Improved insights into the strain-level diversity of complex microbial communities will strengthen our understanding of their role in human health. For example, by comparing the genomes of strains, we could identify genetic factors distinguishing pathogenic and non-pathogenic strains.
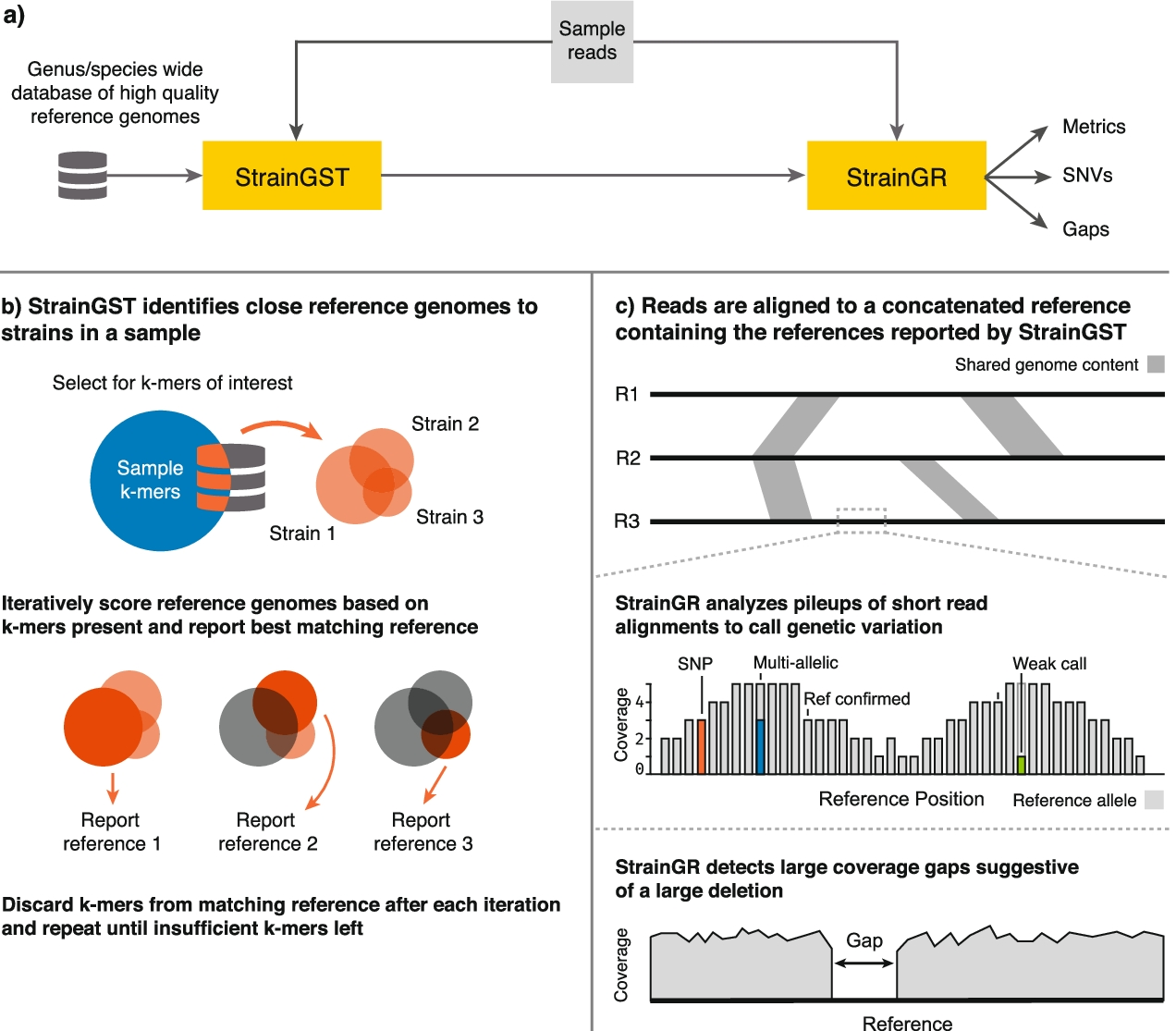
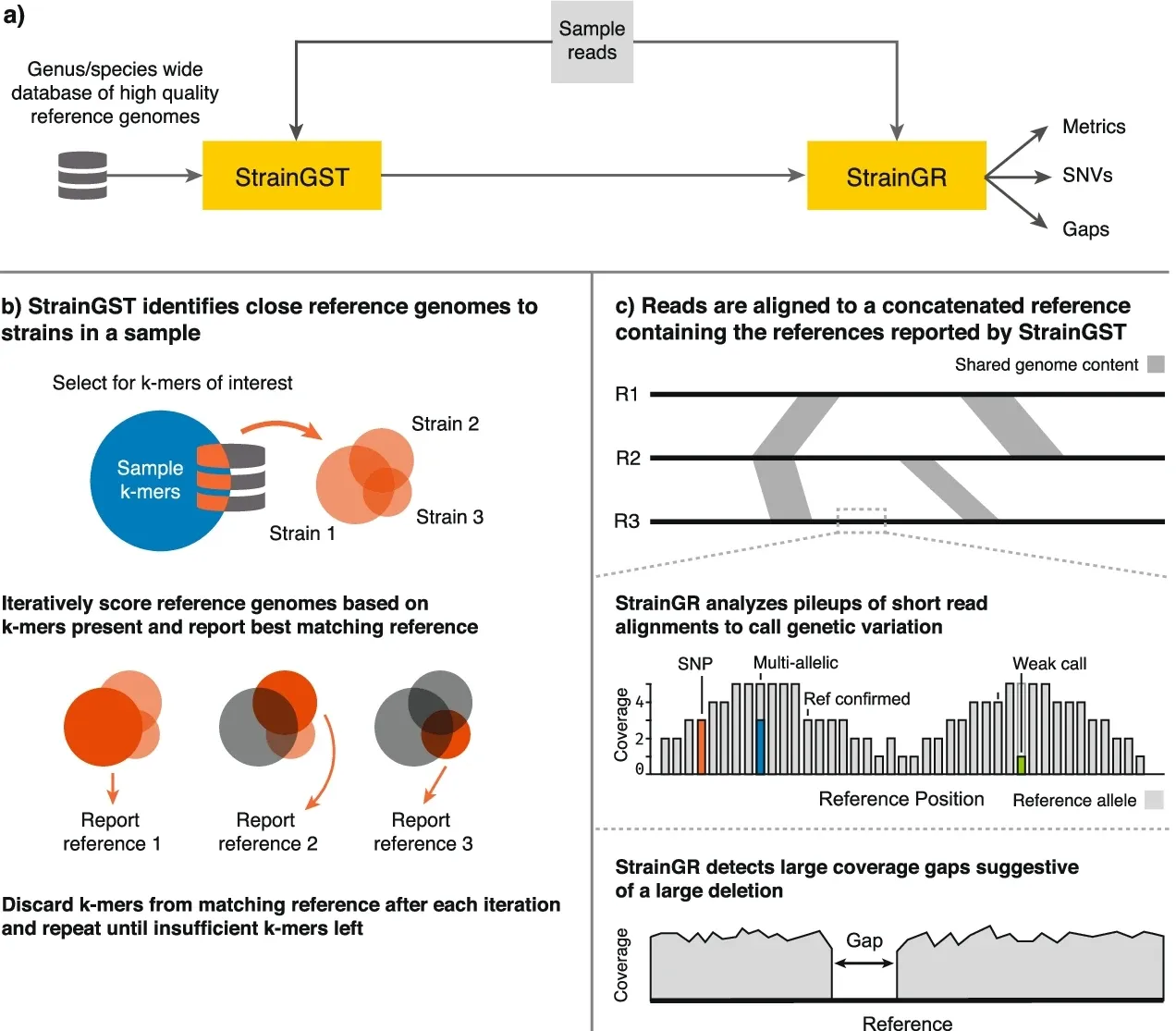
This is where StrainGE comes in. StrainGE is specifically designed to identify and characterize low-abundance strains using metagenomic sequencing data from a community. Metagenomic sequencing data represents sequenced DNA fragments from all community members, which is a challenge for characterizing low-abundance strains. For example, E. coli typically represents only 1% of a healthy human gut microbiome; thus, only a tiny fraction of sequenced reads will originate from E. coli strains.
StrainGE overcomes this challenge by comparing the read data to a database of reference genomes, such as genomes available in public databases. It first detects which reads in the sample likely originate from the species of interest and then compares the reads to the references in the database, reporting those that look the most similar to the strain(s) in the sample. If multiple strains of the same species are present, it will report multiple reference genomes.
StrainGE's reported references serve as a basis for further, more detailed characterization of the sample strains. Since the reported references are unlikely to be the same as the strains in the sample, StrainGE identifies strain-specific genetic variants by mapping sample reads to the references. StrainGE analyzes the read alignment pileups to search for evidence of different alleles compared to the reference.
StrainGE was instrumental in characterizing the E. coli strain-level dynamics in the gut microbiomes of women with recurrent urinary tract infections (UTIs). We found that the UTI-causing strain was rarely cleared from the gut after antibiotics and found unexpected similarities with a healthy control group. More about this study can be found at the link below.